Case Study: Bioreactor Glucose Prediction
Source:vignettes/sec-analysis-example.Rmd
sec-analysis-example.RmdIntroduction
This vignette demonstrates a complete chemometric modeling workflow using measure. We’ll predict glucose concentration in bioreactors using Raman spectroscopy data.
The dataset
The glucose_bioreactors dataset contains Raman spectra
from bioreactor experiments described in Kuhn and Johnson’s Feature
Engineering and Selection (2020).
data(glucose_bioreactors)
# Small-scale bioreactors (15 reactors, 14 days)
dim(bioreactors_small)
#> [1] 210 2655
# Large-scale bioreactors (3 reactors, 14 days)
dim(bioreactors_large)
#> [1] 42 2655Each dataset has: - 2,651 spectral channels (Raman wavenumbers) -
glucose: the target variable to predict - day:
sampling day (1-14) - batch_id and
batch_sample: reactor identifiers
Exploratory visualization
Let’s visualize some spectra from the small-scale reactors:
# Get spectral column names (numeric wavenumbers)
spec_cols <- names(bioreactors_small)[sapply(names(bioreactors_small), function(x) !is.na(suppressWarnings(as.numeric(x))))]
# Convert a few samples to long format for plotting
viz_data <- bioreactors_small |>
slice(1:10) |>
mutate(sample_id = row_number()) |>
select(sample_id, glucose, all_of(spec_cols[1:500])) |> # First 500 channels
pivot_longer(
cols = -c(sample_id, glucose),
names_to = "wavenumber",
values_to = "intensity"
) |>
mutate(wavenumber = as.numeric(wavenumber))
ggplot(viz_data, aes(x = wavenumber, y = intensity, group = sample_id, color = glucose)) +
geom_line(alpha = 0.7) +
scale_color_viridis_c() +
labs(
x = "Wavenumber",
y = "Intensity",
title = "Raman Spectra from Small-Scale Bioreactors",
subtitle = "Color indicates glucose concentration",
color = "Glucose"
) +
theme_minimal()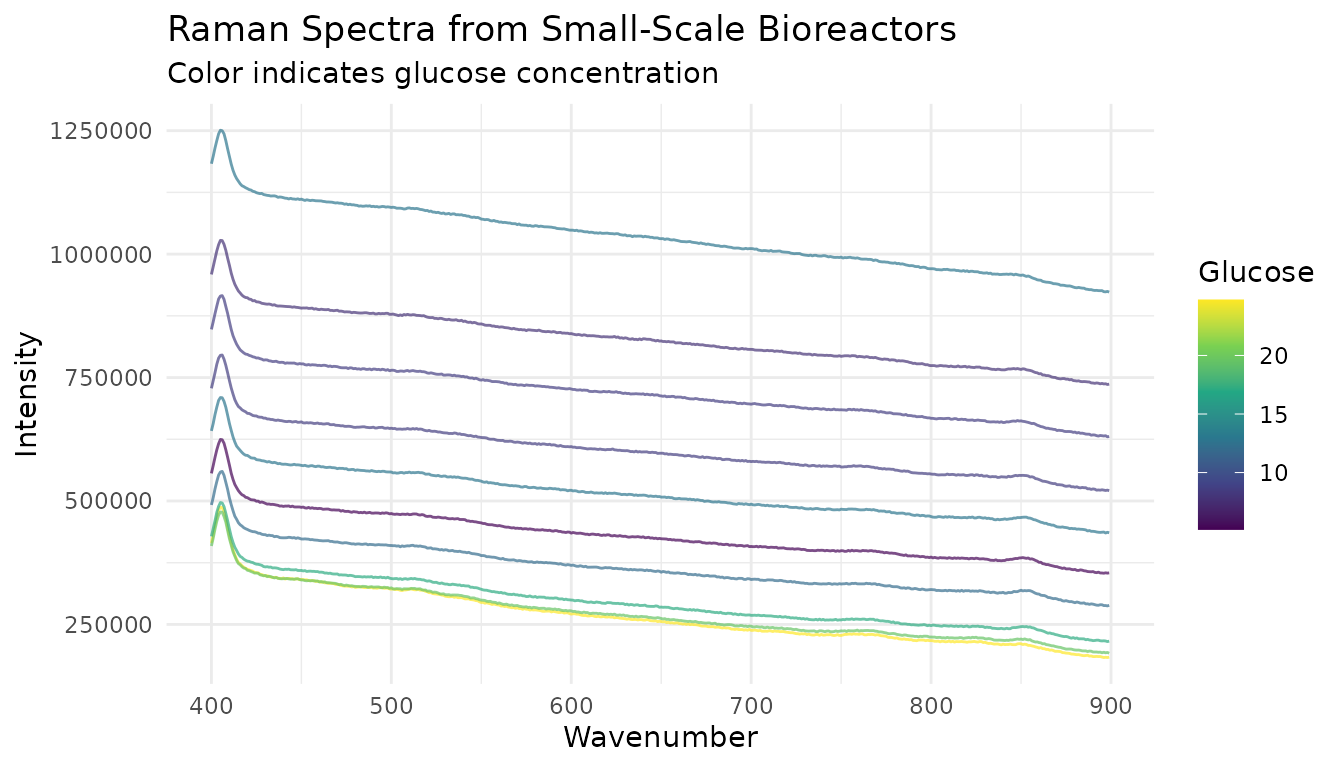
Building a preprocessing recipe
For Raman spectroscopy, a typical preprocessing pipeline includes:
- Smoothing to reduce noise
- Baseline correction (via derivatives)
- Normalization for instrument variations
# Use first 500 spectral columns for faster example
spec_cols_subset <- spec_cols[1:500]
wavenumbers <- as.numeric(spec_cols_subset)
# Prepare training data
train_data <- bioreactors_small |>
select(glucose, day, batch_id, all_of(spec_cols_subset))
# Create preprocessing recipe
rec <- recipe(glucose ~ ., data = train_data) |>
# Don't use day and batch_id as predictors
update_role(day, batch_id, new_role = "id") |>
# Convert wide spectral columns to internal format
step_measure_input_wide(
all_of(spec_cols_subset),
location_values = wavenumbers
) |>
# Savitzky-Golay smoothing + first derivative
step_measure_savitzky_golay(
window_side = 7,
differentiation_order = 1
) |>
# Standard Normal Variate normalization
step_measure_snv() |>
# Convert back to wide format
step_measure_output_wide(prefix = "raman_")
rec
#>
#> ── Recipe ──────────────────────────────────────────────────────────────────────
#>
#> ── Inputs
#> Number of variables by role
#> outcome: 1
#> predictor: 500
#> id: 2
#>
#> ── Operations
#> • Collate wide analytical measurements: all_of(spec_cols_subset)
#> Savitzky-Golay preprocessing on
#> SNV transformation on
#> • Restructure analytical measurements to wide format: "<internal data>"Preparing the data
# Prep the recipe (learns parameters from training data)
rec_prepped <- prep(rec)
# Bake to get preprocessed data
train_processed <- bake(rec_prepped, new_data = NULL)
# Check dimensions
dim(train_processed)
#> [1] 210 489The preprocessed data has fewer columns because: - The
day and batch_id columns are preserved as ID
columns - The 500 spectral columns have been transformed and renamed
with prefix raman_
Visualize preprocessed spectra
# Get preprocessed spectral columns
proc_cols <- names(train_processed)[grepl("^raman_", names(train_processed))]
proc_viz <- train_processed |>
slice(1:10) |>
mutate(sample_id = row_number()) |>
select(sample_id, glucose, all_of(proc_cols)) |>
pivot_longer(
cols = starts_with("raman_"),
names_to = "feature",
values_to = "value"
) |>
mutate(wavenumber = as.numeric(gsub("raman_", "", feature)))
ggplot(proc_viz, aes(x = wavenumber, y = value, group = sample_id, color = glucose)) +
geom_line(alpha = 0.7) +
scale_color_viridis_c() +
labs(
x = "Wavenumber",
y = "Preprocessed Intensity",
title = "Preprocessed Raman Spectra",
subtitle = "After SG derivative + SNV normalization",
color = "Glucose"
) +
theme_minimal()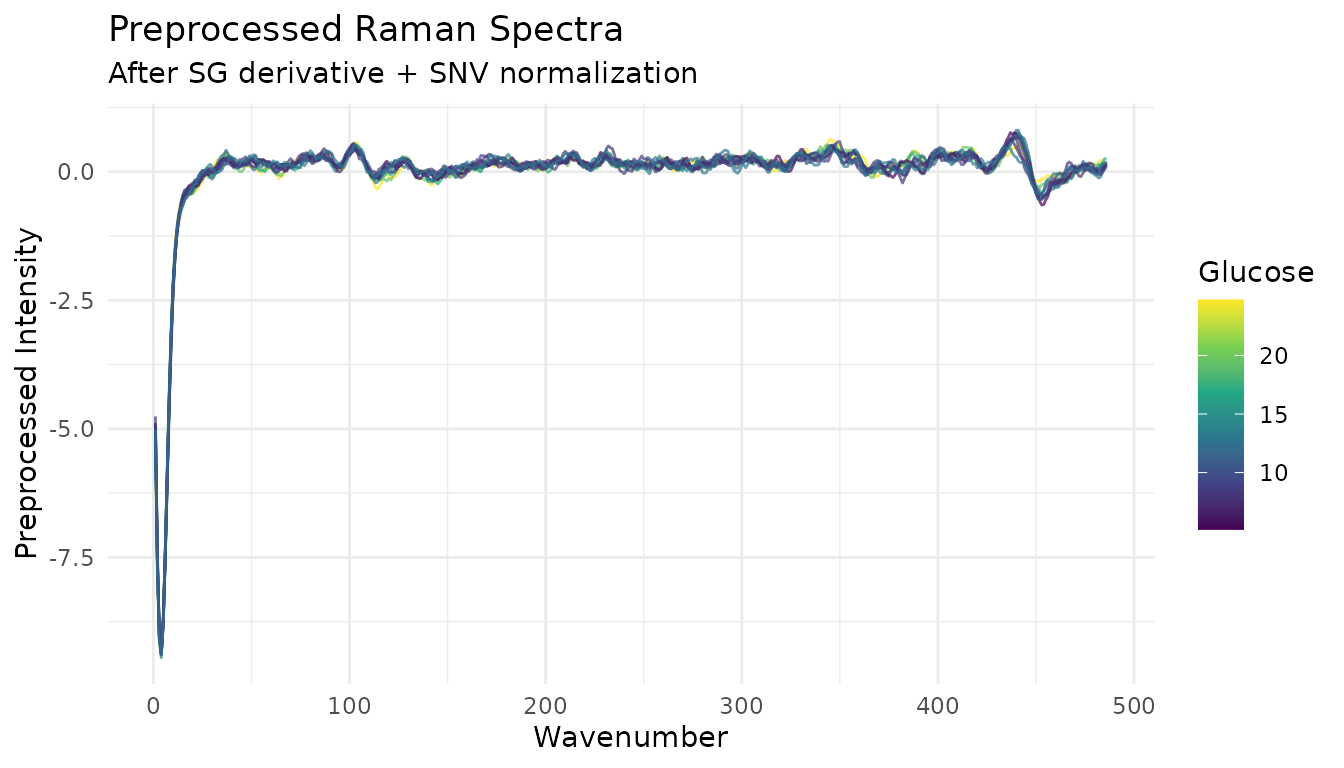
Fitting a model
Now we can fit a model to predict glucose:
library(parsnip)
# Simple linear model (PLS would be better for real applications)
lm_spec <- linear_reg() |>
set_engine("lm")
# Fit model
model <- lm_spec |>
fit(glucose ~ ., data = train_processed |> select(-day, -batch_id))
# Training performance
train_preds <- predict(model, train_processed) |>
bind_cols(train_processed |> select(glucose))
train_preds |>
summarize(
rmse = sqrt(mean((glucose - .pred)^2)),
r_squared = cor(glucose, .pred)^2
)
#> # A tibble: 1 × 2
#> rmse r_squared
#> <dbl> <dbl>
#> 1 1.76e-12 1Applying to new data
The key advantage of the recipes framework is consistent preprocessing of new data:
# Prepare test data from large-scale bioreactors
test_data <- bioreactors_large |>
select(glucose, day, batch_id, all_of(spec_cols_subset))
# Apply the same preprocessing
test_processed <- bake(rec_prepped, new_data = test_data)
# Predict
test_preds <- predict(model, test_processed) |>
bind_cols(test_processed |> select(glucose, batch_id, day))
#> Warning in predict.lm(object = object$fit, newdata = new_data, type =
#> "response", : prediction from rank-deficient fit; consider predict(.,
#> rankdeficient="NA")
# Plot predictions vs actual
ggplot(test_preds, aes(x = glucose, y = .pred, color = batch_id)) +
geom_point(alpha = 0.7) +
geom_abline(slope = 1, intercept = 0, linetype = "dashed") +
labs(
x = "Actual Glucose",
y = "Predicted Glucose",
title = "Model Performance on Large-Scale Bioreactors",
color = "Reactor"
) +
theme_minimal()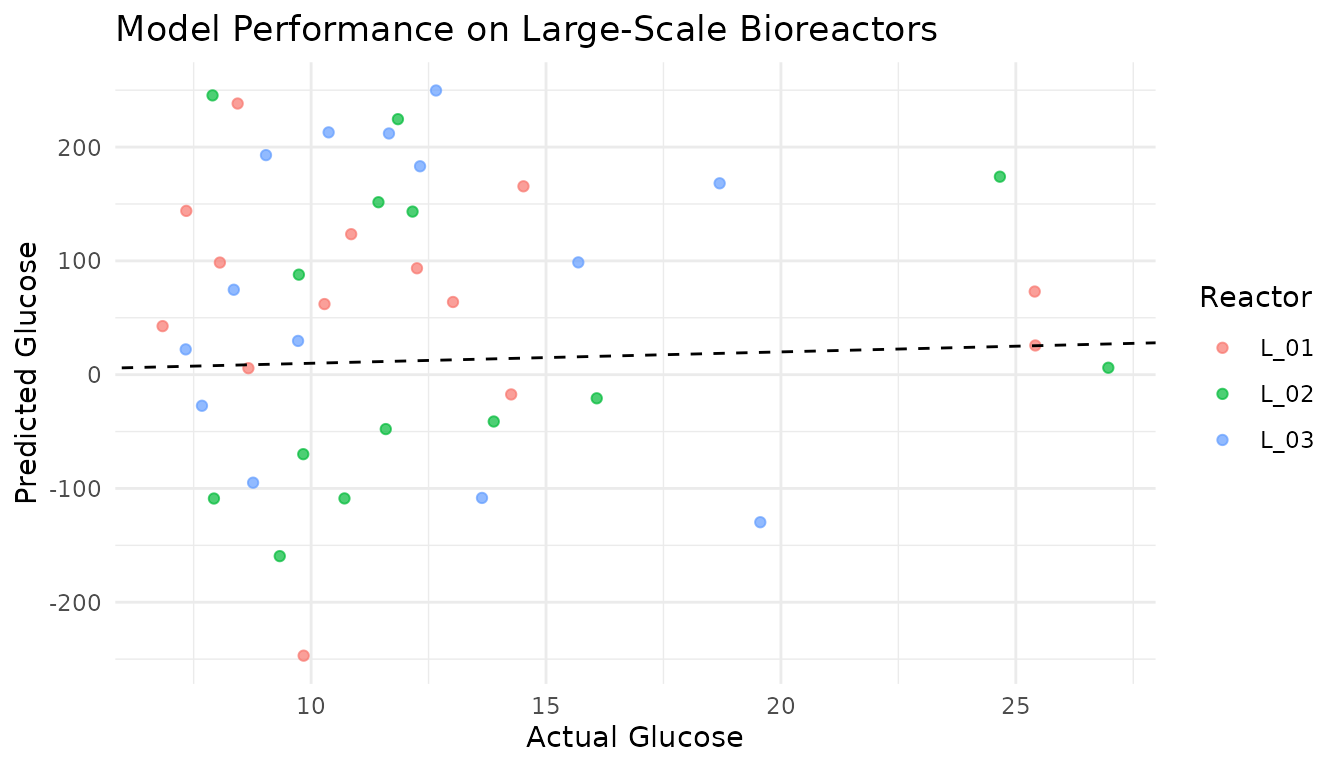
Summary
This case study demonstrated:
- Loading and exploring spectral data from bioreactors
- Building a preprocessing recipe with measure steps
- Fitting a model on preprocessed training data
- Applying consistent preprocessing to new data
Key takeaways:
- The recipe framework ensures identical preprocessing for training and test data
-
step_measure_input_wide()handles the conversion of many spectral columns - Processing steps like Savitzky-Golay and SNV remove unwanted variation
-
step_measure_output_wide()produces modeling-ready data
For real applications, consider:
- Using PLS or ridge regression for high-dimensional spectral data
- Tuning preprocessing parameters with cross-validation
- Validating on truly independent test sets