Data Organization and Diagnostic Plots
Source:vignettes/roles-diagnostics.Rmd
roles-diagnostics.RmdIntroduction
When working with analytical data, proper organization and visualization are essential for building effective preprocessing pipelines. The measure package provides tools to:
- Detect column types automatically based on naming conventions
- Assign roles to columns for use in recipes
- Validate recipes before running them
- Visualize spectra and preprocessing effects
This vignette covers these data organization and diagnostic capabilities.
Detecting Column Types
Analytical data often follows naming conventions that indicate what
each column represents. The measure_identify_columns()
function automatically detects these patterns:
| Prefix | Type | Description |
|---|---|---|
wn_* |
wavenumber | IR spectroscopy (cm⁻¹) |
nm_* |
wavelength | UV-Vis, NIR spectroscopy |
rt_* |
retention_time | Chromatography |
mz_* |
mz | Mass spectrometry |
ppm_* |
ppm | NMR chemical shift |
ch_* |
channel | Generic channel data |
x_* |
generic | Generic measurements |
Example: Identifying columns in wide-format data
# Create example wide-format spectral data
spec_data <- data.frame(
sample_id = paste0("S", 1:5),
concentration = c(10.2, 25.1, 50.3, 75.0, 100.5),
batch = c("A", "A", "B", "B", "B"),
wn_1000 = rnorm(5),
wn_1001 = rnorm(5),
wn_1002 = rnorm(5),
wn_1003 = rnorm(5),
wn_1004 = rnorm(5)
)
# Identify column types
col_info <- measure_identify_columns(spec_data)
col_info
#> # A tibble: 8 × 5
#> column type suggested_role n_values class
#> <chr> <chr> <chr> <int> <chr>
#> 1 sample_id other id 5 character
#> 2 concentration other outcome 5 numeric
#> 3 batch other predictor 5 character
#> 4 wn_1000 wavenumber predictor 5 numeric
#> 5 wn_1001 wavenumber predictor 5 numeric
#> 6 wn_1002 wavenumber predictor 5 numeric
#> 7 wn_1003 wavenumber predictor 5 numeric
#> 8 wn_1004 wavenumber predictor 5 numericThe function returns a tibble with: - column: Column name - type: Detected type (or “other” if no pattern matched) - suggested_role: Recommended recipe role - n_values: Count of non-NA values - class: R class of the column
Summarizing by type
For a quick overview, use measure_column_summary():
measure_column_summary(spec_data)
#> # A tibble: 2 × 3
#> type n_columns example_cols
#> <chr> <int> <chr>
#> 1 wavenumber 5 wn_1000, wn_1001, wn_1002
#> 2 other 3 sample_id, concentration, batchAssigning Roles in Recipes
Recipes use roles to determine how columns should be treated. Common roles include:
| Role | Purpose |
|---|---|
predictor |
Used as model input |
outcome |
Target variable for modeling |
id |
Sample identifier (excluded from modeling) |
blank |
Blank/background samples |
qc |
Quality control samples |
standard |
Calibration standards |
metadata |
Sample metadata (excluded from modeling) |
Using set_measure_roles()
The set_measure_roles() function provides a convenient
way to assign multiple roles at once:
rec <- recipe(concentration ~ ., data = spec_data) |>
set_measure_roles(
id_cols = sample_id,
metadata_cols = batch
)
# Check the assigned roles
rec$var_info
#> # A tibble: 8 × 4
#> variable type role source
#> <chr> <list> <chr> <chr>
#> 1 sample_id <chr [3]> id original
#> 2 batch <chr [3]> metadata original
#> 3 wn_1000 <chr [2]> predictor original
#> 4 wn_1001 <chr [2]> predictor original
#> 5 wn_1002 <chr [2]> predictor original
#> 6 wn_1003 <chr [2]> predictor original
#> 7 wn_1004 <chr [2]> predictor original
#> 8 concentration <chr [2]> outcome originalThis is equivalent to calling update_role() multiple
times, but more concise for common analytical data patterns.
Custom roles for analytical workflows
For analytical chemistry workflows, you might want to identify special sample types:
# Example with QC and blank samples
analytical_data <- data.frame(
sample_id = c("blank_1", "QC_1", "S1", "S2", "QC_2", "S3"),
sample_type = c("blank", "qc", "sample", "sample", "qc", "sample"),
concentration = c(NA, 50, 10, 25, 50, 75),
wn_1000 = rnorm(6),
wn_1001 = rnorm(6)
)
rec <- recipe(concentration ~ ., data = analytical_data) |>
set_measure_roles(
id_cols = sample_id,
blank_cols = starts_with("blank"),
qc_cols = starts_with("QC")
)Validating Recipe Structure
Before running a preprocessing pipeline,
check_measure_recipe() validates the recipe structure and
identifies potential issues:
# A well-structured recipe
data(meats_long)
good_rec <- recipe(water + fat + protein ~ ., data = meats_long) |>
update_role(id, new_role = "id") |>
step_measure_input_long(transmittance, location = vars(channel)) |>
step_measure_snv() |>
step_measure_output_wide()
issues <- check_measure_recipe(good_rec)
issues
#> # A tibble: 0 × 3
#> # ℹ 3 variables: level <chr>, check <chr>, message <chr>Detecting common issues
The function checks for:
Errors (will cause failures): - No input step - Multiple input steps - Output step before input step
Warnings (may cause issues): - No output step - Processing steps after output step
Info (suggestions): - No ID column - Large number of predictors
# A recipe with issues
bad_rec <- recipe(water ~ ., data = meats_long) |>
step_measure_snv() # Missing input step!
issues <- check_measure_recipe(bad_rec)
issues
#> # A tibble: 3 × 3
#> level check message
#> <chr> <chr> <chr>
#> 1 error no_input Recipe has no input step. Add step_measure_input_wide() or …
#> 2 warning no_output Recipe has no output step. Data will remain in internal .me…
#> 3 info no_id No ID column identified. Consider using update_role(col, ne…Interactive mode
Use strict = FALSE for interactive feedback:
check_measure_recipe(bad_rec, strict = FALSE)
# ✖ Recipe has no input step. Add step_measure_input_wide() or step_measure_input_long().Visualizing Spectra
The measure package provides autoplot() methods for
quick visualization of spectral data.
Plotting a single spectrum
# Create a single spectrum
spec <- new_measure_tbl(
location = seq(1000, 1100, by = 1),
value = sin(seq(1000, 1100, by = 1) / 20) + rnorm(101, sd = 0.1)
)
autoplot(spec)
Plotting multiple spectra
# Process some data to get a measure_list
rec <- recipe(water + fat + protein ~ ., data = meats_long) |>
update_role(id, new_role = "id") |>
step_measure_input_long(transmittance, location = vars(channel)) |>
prep(retain = TRUE)
baked <- bake(rec, new_data = NULL)
# Plot the spectra
autoplot(baked$.measures, max_spectra = 20)
Adding summary statistics
Use summary = TRUE to overlay mean ± standard
deviation:
autoplot(baked$.measures, summary = TRUE, max_spectra = 30, alpha = 0.2)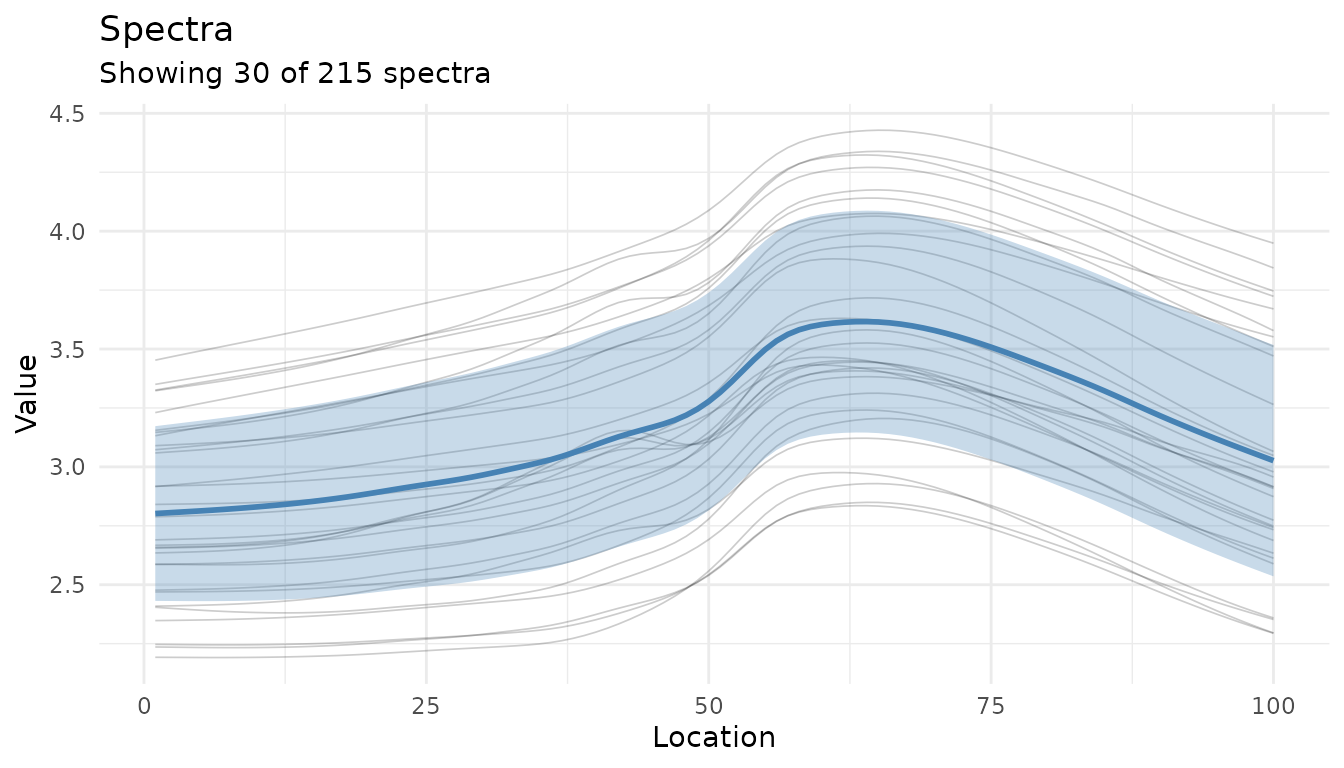
Visualizing Preprocessing Effects
Before/after comparison
The autoplot() method for recipes shows preprocessing
effects:
# Create a preprocessing recipe
rec <- recipe(water + fat + protein ~ ., data = meats_long) |>
update_role(id, new_role = "id") |>
step_measure_input_long(transmittance, location = vars(channel)) |>
step_measure_savitzky_golay(window_side = 5, differentiation_order = 1) |>
step_measure_snv() |>
prep(retain = TRUE)
autoplot(rec, n_samples = 10)
#> Warning: Could not extract 'before' data for comparison.
#> ℹ Showing processed data only.
#> ✖ ℹ In argument: `dplyr::all_of(rename_map)`. Caused by error in
#> `dplyr::all_of()`: ! Can't subset elements that don't exist. ✖ Elements
#> `transmittance` and `channel` don't exist.
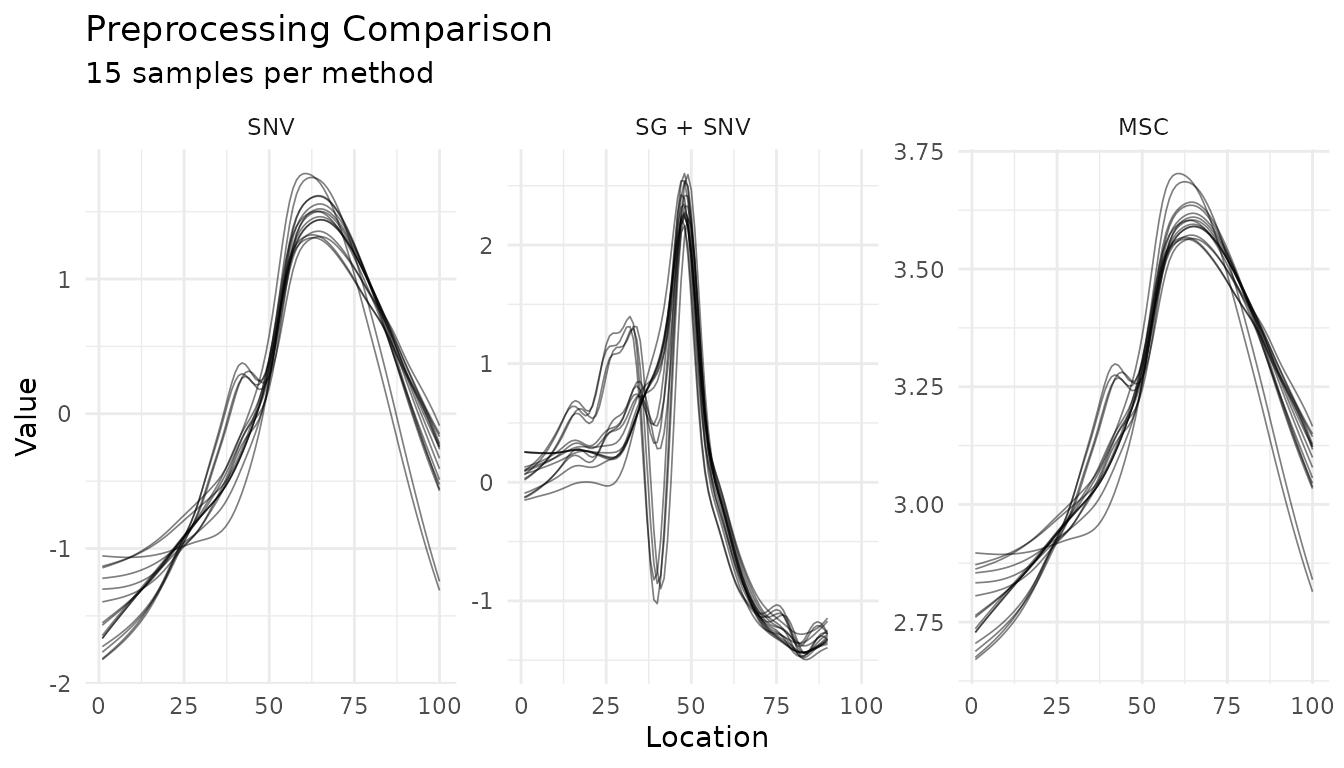
Comparing Preprocessing Strategies
Use plot_measure_comparison() to compare different
preprocessing approaches side-by-side:
# Define different preprocessing strategies
base_rec <- recipe(water + fat + protein ~ ., data = meats_long) |>
update_role(id, new_role = "id") |>
step_measure_input_long(transmittance, location = vars(channel))
# Strategy 1: Just SNV
snv_rec <- base_rec |>
step_measure_snv() |>
prep(retain = TRUE)
# Strategy 2: Savitzky-Golay + SNV
sg_snv_rec <- base_rec |>
step_measure_savitzky_golay(window_side = 5, differentiation_order = 1) |>
step_measure_snv() |>
prep(retain = TRUE)
# Strategy 3: MSC
msc_rec <- base_rec |>
step_measure_msc() |>
prep(retain = TRUE)
# Compare all three
plot_measure_comparison(
"SNV" = snv_rec,
"SG + SNV" = sg_snv_rec,
"MSC" = msc_rec,
n_samples = 15
)
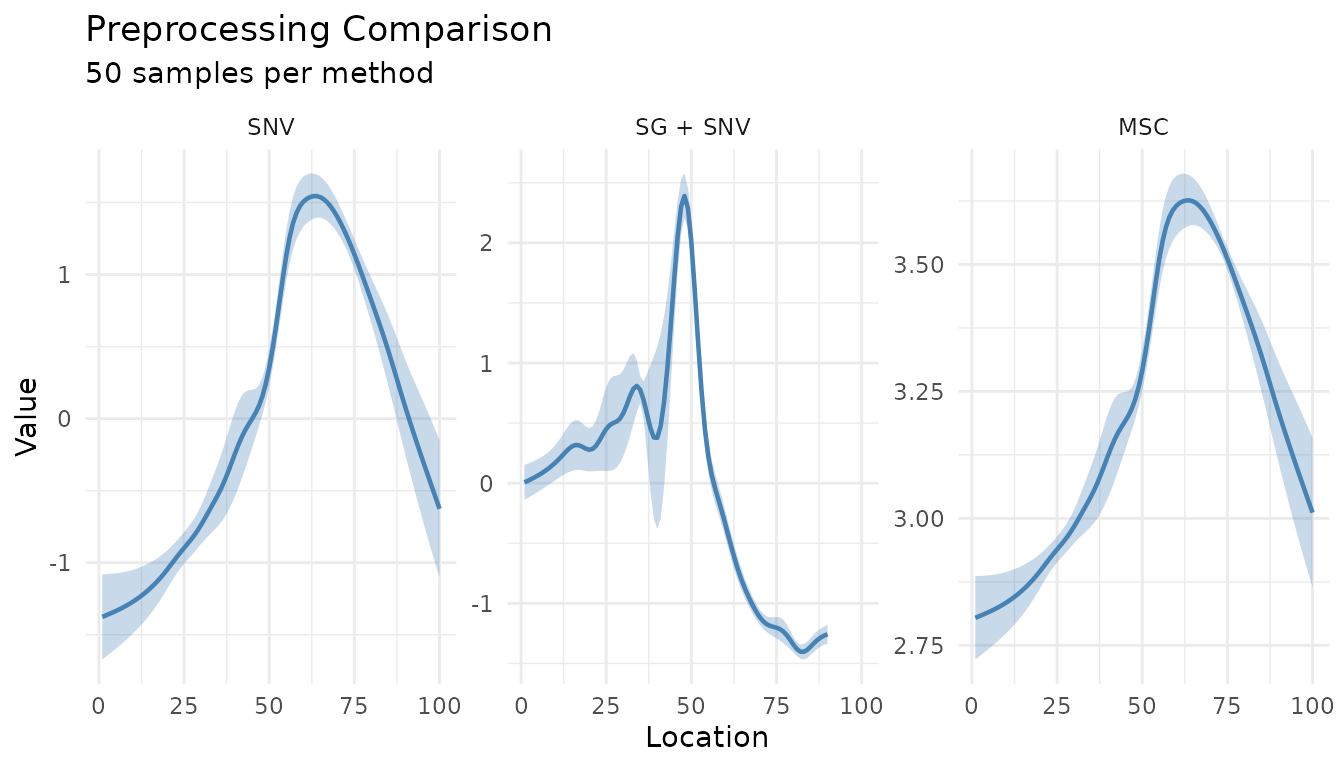
Summary comparison
For a cleaner comparison, use summary_only = TRUE:
plot_measure_comparison(
"SNV" = snv_rec,
"SG + SNV" = sg_snv_rec,
"MSC" = msc_rec,
n_samples = 50,
summary_only = TRUE
)
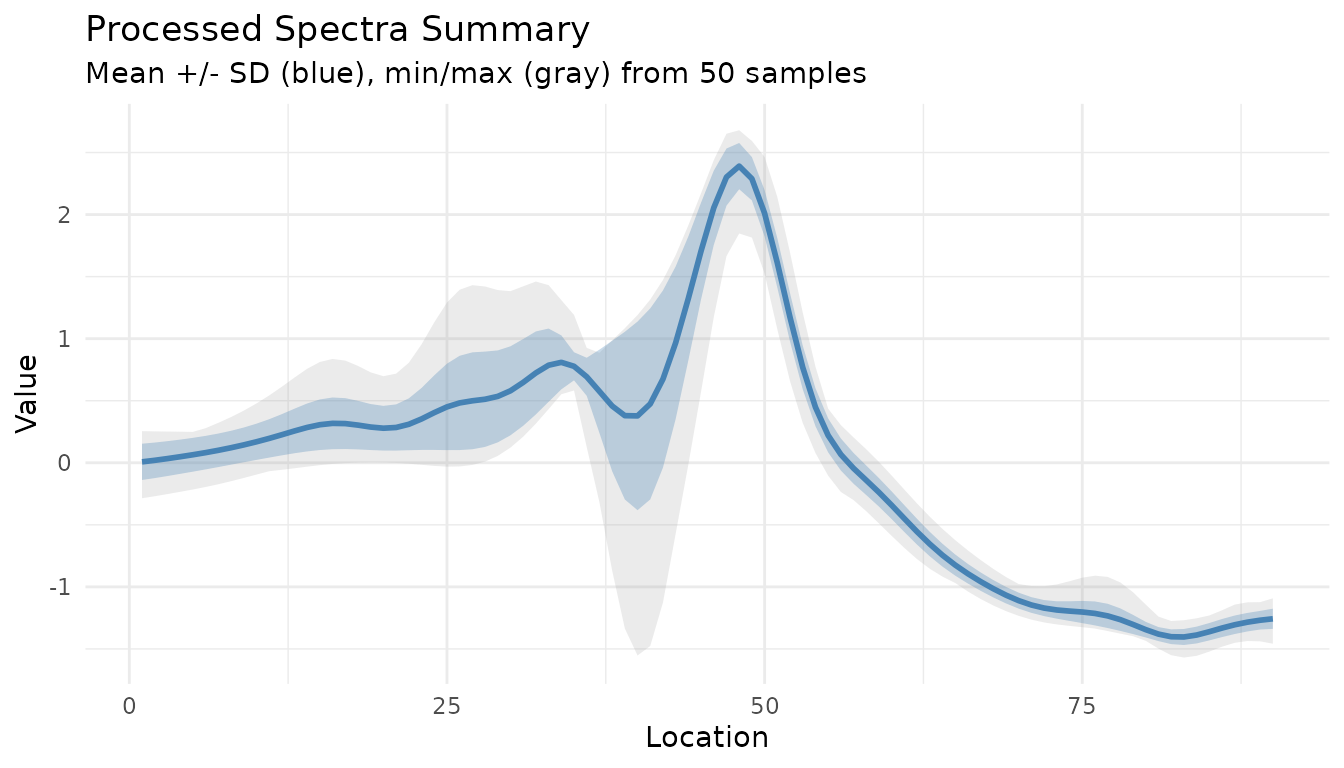
Summary Plot for Processed Data
The measure_plot_summary() function creates
publication-ready summary plots:
baked <- bake(sg_snv_rec, new_data = NULL)
measure_plot_summary(baked)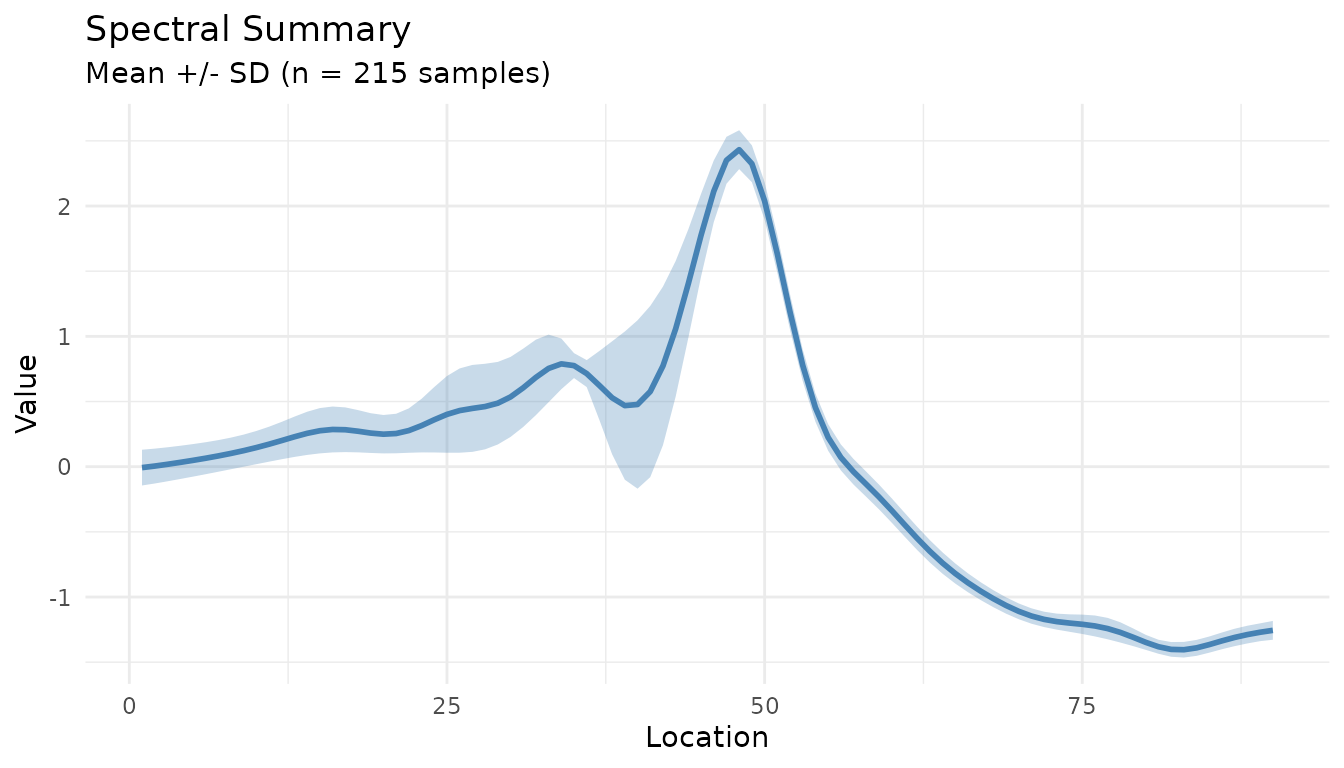
Show the full range with show_range = TRUE:
measure_plot_summary(baked, show_range = TRUE)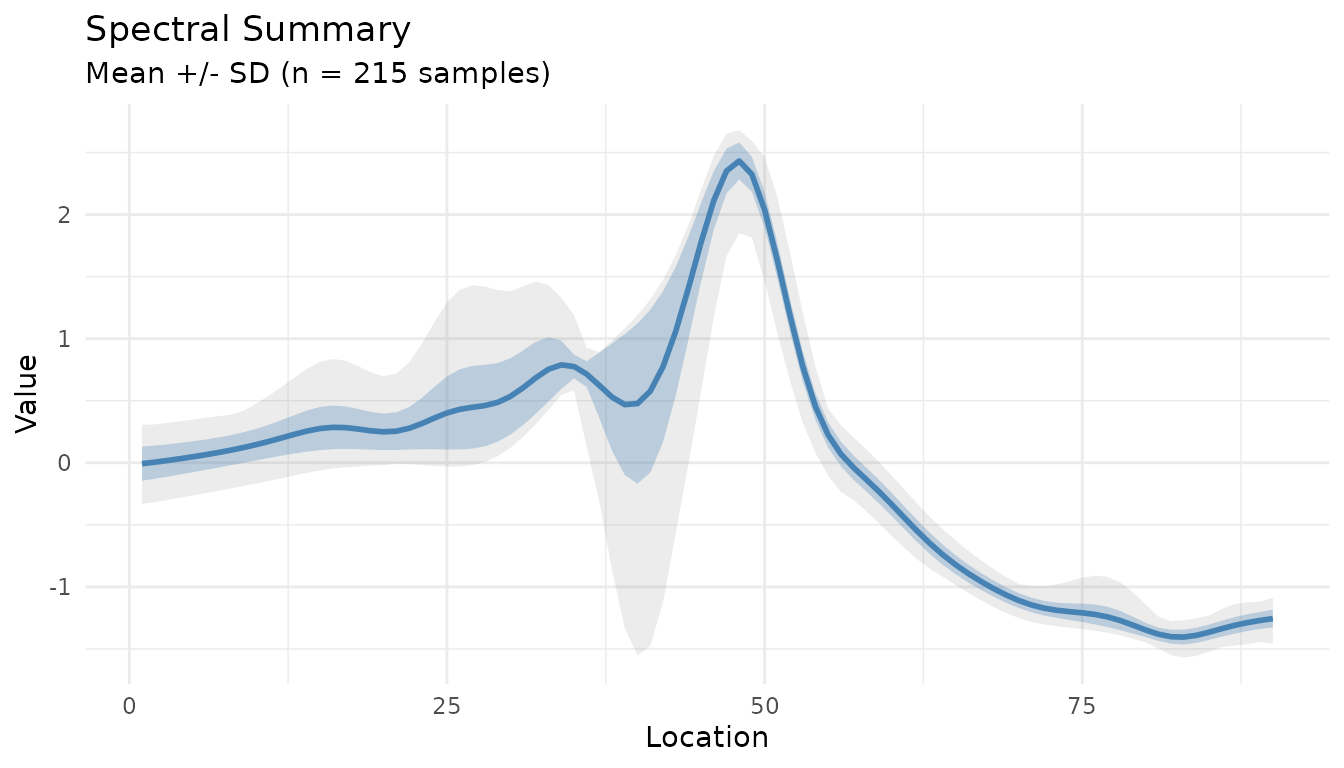
Best Practices
Always check your recipe with
check_measure_recipe()before running long preprocessing pipelinesUse
measure_identify_columns()to understand your data structure before building recipesAssign roles explicitly for ID columns, metadata, and special sample types
Visualize at each stage - use
autoplot()to verify preprocessing effectsCompare strategies with
plot_measure_comparison()before committing to a preprocessing approach
Next Steps
- See
vignette("preprocessing")for details on all preprocessing steps - See
vignette("recipes")for integration with tidymodels workflows - Explore hyperparameter tuning for preprocessing steps with
tune