Introduction
Baseline correction is a fundamental preprocessing step for spectroscopic and chromatographic data. Baselines can drift due to instrument effects, sample scattering, fluorescence, or detector response, obscuring the chemical information in your measurements.
The measure package provides several baseline correction methods as recipe steps, plus the ability to use custom R functions or Python’s pybaselines library.
The problem: Baseline drift
Let’s visualize baseline issues in NIR spectra:
data(meats)
# Convert to long format for visualization
meats_viz <- meats |>
mutate(id = row_number()) |>
pivot_longer(
cols = starts_with("x_"),
names_to = "channel",
values_to = "transmittance"
) |>
mutate(channel = as.integer(gsub("x_", "", channel)))
# Plot raw spectra
meats_viz |>
filter(id <= 20) |>
ggplot(aes(x = channel, y = transmittance, group = id, color = factor(id))) +
geom_line(alpha = 0.7) +
labs(
x = "Channel",
y = "Transmittance",
title = "Raw NIR Spectra with Baseline Variation"
) +
theme_minimal() +
theme(legend.position = "none")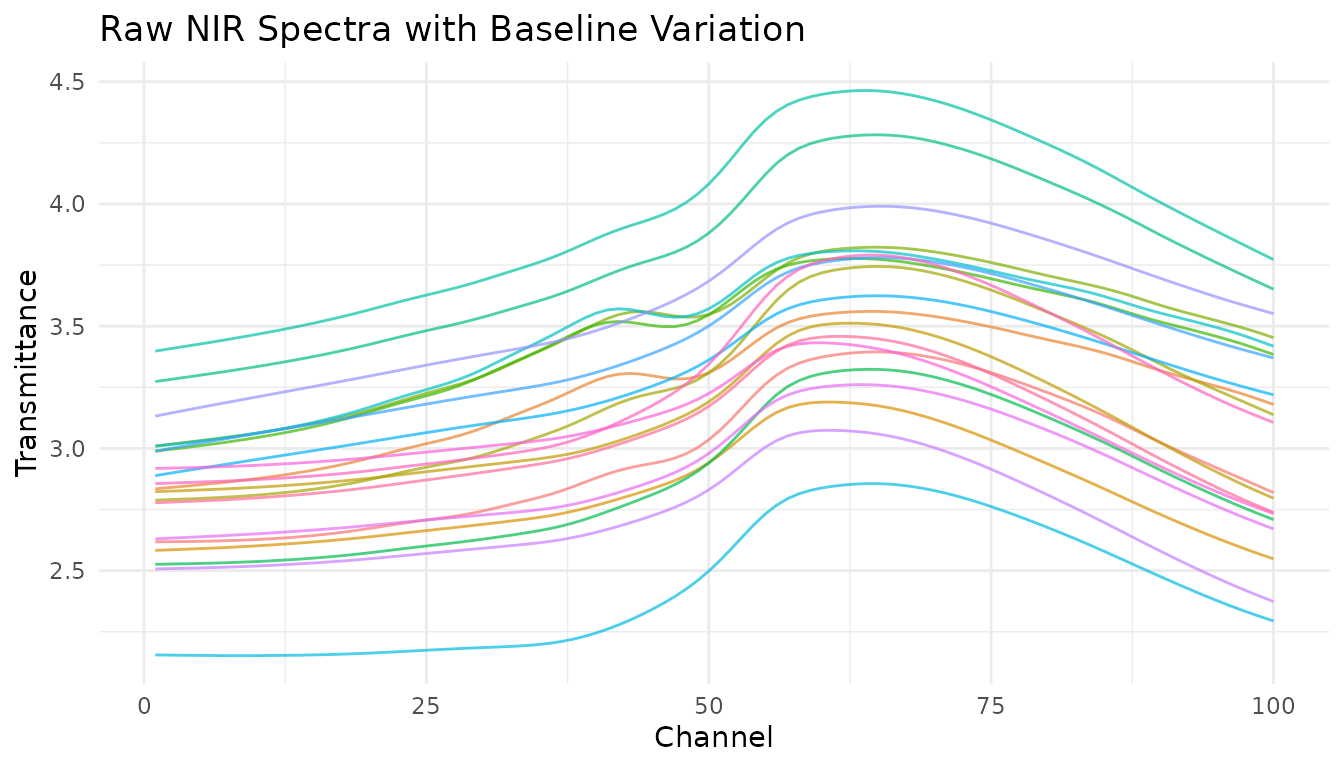
Notice the vertical offset between spectra? This baseline shift isn’t related to the chemical composition we want to model.
Built-in baseline correction methods
Asymmetric Least Squares (ALS)
step_measure_baseline_als() uses the Asymmetric Least
Squares algorithm, which is excellent for spectra where peaks are
predominantly in one direction (e.g., absorption peaks going up or
emission peaks going down).
rec_als <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_als(lambda = 1e6, p = 0.01)
processed_als <- bake(prep(rec_als), new_data = NULL)
# Visualize using unnest
plot_als <- processed_als |>
slice(1:20) |>
mutate(id = row_number()) |>
unnest(.measures)
ggplot(plot_als, aes(x = location, y = value, group = id, color = factor(id))) +
geom_line(alpha = 0.7) +
labs(
x = "Channel",
y = "Corrected Transmittance",
title = "ALS Baseline Correction"
) +
theme_minimal() +
theme(legend.position = "none")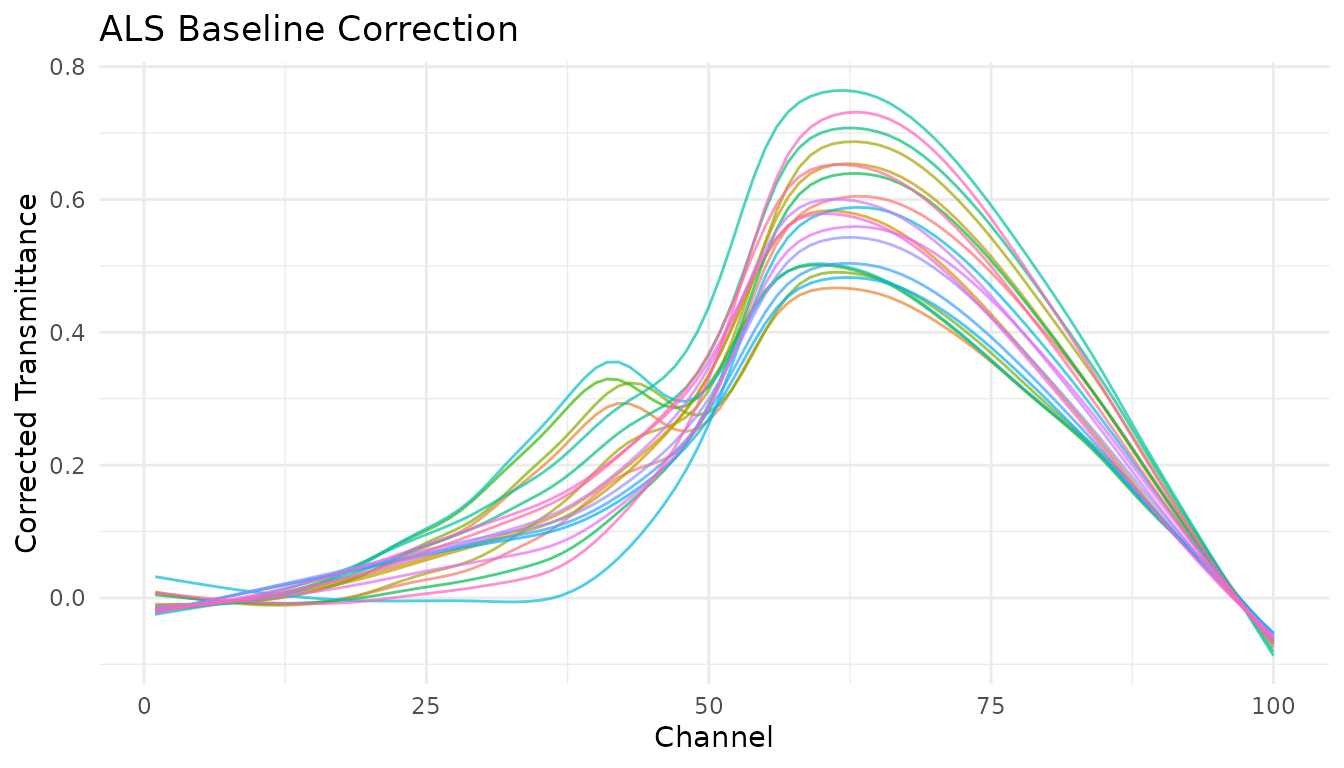
Key parameters:
-
lambda: Smoothness penalty (higher = smoother baseline). Try 10^4 to 10^9. -
p: Asymmetry parameter (0-1). Lower values fit below the signal. Try 0.001-0.1.
Polynomial baseline
step_measure_baseline_poly() fits a polynomial to the
spectrum and subtracts it. Simple and fast, works well for gentle
baseline curvature.
rec_poly <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_poly(degree = 2)
processed_poly <- bake(prep(rec_poly), new_data = NULL)
# Visualize using unnest
plot_poly <- processed_poly |>
slice(1:20) |>
mutate(id = row_number()) |>
unnest(.measures)
ggplot(plot_poly, aes(x = location, y = value, group = id, color = factor(id))) +
geom_line(alpha = 0.7) +
labs(
x = "Channel",
y = "Corrected Transmittance",
title = "Polynomial Baseline Correction (degree = 2)"
) +
theme_minimal() +
theme(legend.position = "none")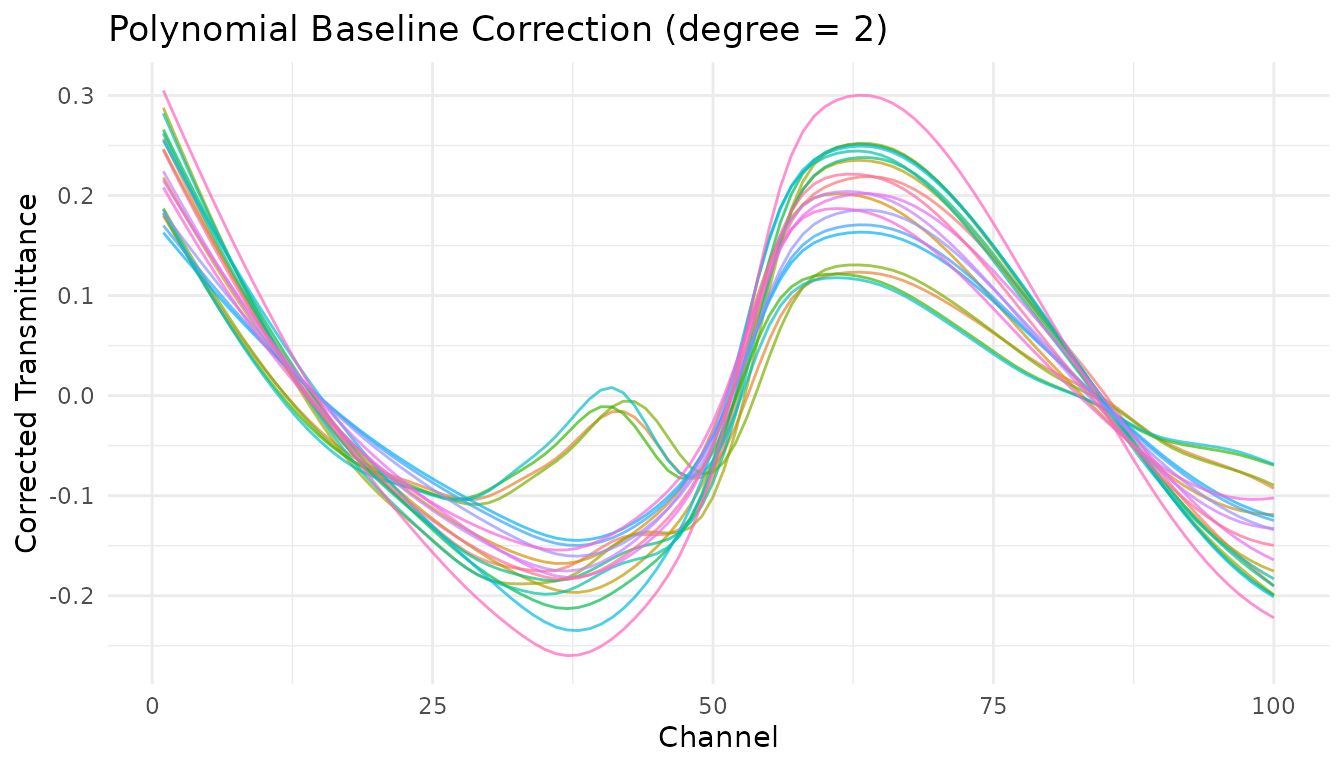
Robust fitting baseline
step_measure_baseline_rf() uses robust local regression
(LOESS with iterative reweighting) to estimate the baseline. Good for
complex baseline shapes.
rec_rf <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_rf(span = 0.5) |>
step_measure_output_wide(prefix = "nir_")
processed_rf <- bake(prep(rec_rf), new_data = NULL)
#> Warning: Values from `value` are not uniquely identified; output will contain list-cols.
#> • Use `values_fn = list` to suppress this warning.
#> • Use `values_fn = {summary_fun}` to summarise duplicates.
#> • Use the following dplyr code to identify duplicates.
#> {data} |>
#> dplyr::summarise(n = dplyr::n(), .by = c(water, fat, protein, location)) |>
#> dplyr::filter(n > 1L)Detrending
step_measure_detrend() removes polynomial trends from
spectra. The simplest baseline correction - just removes the overall
trend.
rec_detrend <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_detrend(degree = 1) |>
step_measure_output_wide(prefix = "nir_")
processed_detrend <- bake(prep(rec_detrend), new_data = NULL)
#> Warning: Values from `value` are not uniquely identified; output will contain list-cols.
#> • Use `values_fn = list` to suppress this warning.
#> • Use `values_fn = {summary_fun}` to summarise duplicates.
#> • Use the following dplyr code to identify duplicates.
#> {data} |>
#> dplyr::summarise(n = dplyr::n(), .by = c(water, fat, protein, location)) |>
#> dplyr::filter(n > 1L)GPC/SEC baseline correction
step_measure_baseline_gpc() is specialized for
chromatography data where baseline regions exist at the start and end of
the chromatogram.
# For chromatography data
rec_gpc <- recipe(outcome ~ ., data = chromatogram_data) |>
step_measure_input_long(signal, location = vars(time)) |>
step_measure_baseline_gpc(left_frac = 0.05, right_frac = 0.05, method = "linear")Custom baseline functions
step_measure_baseline_custom() lets you provide any R
function for baseline estimation. Your function receives a
measure_tbl (tibble with location and
value columns) and should return a numeric vector of
baseline values.
Using a function
# Simple moving minimum baseline
moving_min_baseline <- function(x, window = 51) {
y <- x$value
n <- length(y)
baseline <- numeric(n)
half_win <- window %/% 2
for (i in seq_len(n)) {
start <- max(1, i - half_win)
end <- min(n, i + half_win)
baseline[i] <- min(y[start:end], na.rm = TRUE)
}
baseline
}
rec_custom <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_custom(.fn = moving_min_baseline, window = 101) |>
step_measure_output_wide(prefix = "nir_")
processed_custom <- bake(prep(rec_custom), new_data = NULL)
#> Warning: Values from `value` are not uniquely identified; output will contain list-cols.
#> • Use `values_fn = list` to suppress this warning.
#> • Use `values_fn = {summary_fun}` to summarise duplicates.
#> • Use the following dplyr code to identify duplicates.
#> {data} |>
#> dplyr::summarise(n = dplyr::n(), .by = c(water, fat, protein, location)) |>
#> dplyr::filter(n > 1L)Using a formula
For quick one-liners, use the formula interface where .x
is the measure_tbl:
# LOESS baseline using formula
rec_loess <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_custom(
.fn = ~ stats::loess(.x$value ~ .x$location, span = span)$fitted,
span = 0.3
) |>
step_measure_output_wide(prefix = "nir_")
processed_loess <- bake(prep(rec_loess), new_data = NULL)
#> Warning: Values from `value` are not uniquely identified; output will contain list-cols.
#> • Use `values_fn = list` to suppress this warning.
#> • Use `values_fn = {summary_fun}` to summarise duplicates.
#> • Use the following dplyr code to identify duplicates.
#> {data} |>
#> dplyr::summarise(n = dplyr::n(), .by = c(water, fat, protein, location)) |>
#> dplyr::filter(n > 1L)Extracting the baseline
With step_measure_baseline_custom() and
step_measure_baseline_py(), set
subtract = FALSE to get the baseline itself instead of the
corrected signal:
# Use custom baseline with subtract = FALSE to extract the baseline
rec_extract <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_custom(
.fn = ~ stats::loess(.x$value ~ .x$location, span = 0.5)$fitted,
subtract = FALSE
)
baselines <- bake(prep(rec_extract), new_data = NULL)
# The .measures column now contains the estimated baselines
baselines |>
slice(1) |>
unnest(.measures) |>
head()
#> # A tibble: 6 × 5
#> water fat protein location value
#> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 60.5 22.5 16.7 1 2.62
#> 2 60.5 22.5 16.7 2 2.62
#> 3 60.5 22.5 16.7 3 2.62
#> 4 60.5 22.5 16.7 4 2.62
#> 5 60.5 22.5 16.7 5 2.62
#> 6 60.5 22.5 16.7 6 2.62Python pybaselines integration
step_measure_baseline_py() provides access to over 50
baseline correction algorithms from the Python pybaselines library.
Setup
First, install pybaselines:
# Install reticulate if needed
install.packages("reticulate")
# Install pybaselines
reticulate::py_require("pybaselines")Using pybaselines methods
# Asymmetric Least Squares
rec_py_asls <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_py(method = "asls", lam = 1e6, p = 0.01) |>
step_measure_output_wide(prefix = "nir_")
# SNIP algorithm (good for spectroscopy)
rec_py_snip <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_py(method = "snip", max_half_window = 40) |>
step_measure_output_wide(prefix = "nir_")
# Modified polynomial
rec_py_modpoly <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_py(method = "modpoly", poly_order = 3) |>
step_measure_output_wide(prefix = "nir_")
# Morphological (rolling ball)
rec_py_mor <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_py(method = "rolling_ball", half_window = 30) |>
step_measure_output_wide(prefix = "nir_")Available pybaselines methods
| Category | Methods |
|---|---|
| Whittaker |
asls, iasls, airpls,
arpls, drpls, psalsa
|
| Polynomial |
poly, modpoly, imodpoly,
loess, quant_reg
|
| Morphological |
mor, imor, rolling_ball,
tophat
|
| Spline |
pspline_asls, pspline_airpls,
mixture_model
|
| Smooth |
snip, swima,
noise_median
|
| Classification |
dietrich, golotvin,
fastchrom
|
See the pybaselines documentation for the full list and parameter details.
Using derivatives for baseline correction
Savitzky-Golay derivatives remain a powerful approach for baseline correction, especially in NIR spectroscopy:
# First derivative removes constant baseline
rec_d1 <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_savitzky_golay(window_side = 7, differentiation_order = 1) |>
step_measure_output_wide(prefix = "nir_")
# Second derivative removes linear baseline
rec_d2 <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_savitzky_golay(window_side = 7, differentiation_order = 2) |>
step_measure_output_wide(prefix = "nir_")Choosing a method
| Method | Best for | Tunable | Notes |
|---|---|---|---|
step_measure_baseline_als() |
Most spectroscopy | Yes | Good general-purpose choice |
step_measure_baseline_poly() |
Gentle curvature | Yes | Simple and fast |
step_measure_baseline_rf() |
Complex baselines | Yes | Robust to outliers |
step_measure_detrend() |
Simple trends | Yes | Fastest option |
step_measure_baseline_gpc() |
Chromatography | No | Uses baseline regions |
step_measure_baseline_custom() |
Special cases | Optional | Maximum flexibility |
step_measure_baseline_py() |
Advanced algorithms | Yes | 50+ methods available |
step_measure_savitzky_golay() |
NIR/IR spectra | Yes | Also smooths and differentiates |
Hyperparameter tuning
Most baseline correction steps are tunable with the tidymodels tuning framework:
library(tune)
# Create tunable recipe
rec_tunable <- recipe(water ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_als(
lambda = tune(), # Will be tuned
p = tune() # Will be tuned
) |>
step_measure_output_wide(prefix = "nir_")
# The tunable parameters are automatically detected
tunable(rec_tunable)For step_measure_baseline_custom(), you can declare
tunable parameters explicitly:
rec_custom_tunable <- recipe(water ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
step_measure_baseline_custom(
.fn = ~ stats::loess(.x$value ~ .x$location, span = span)$fitted,
span = 0.5,
tunable = list(
span = list(pkg = "dials", fun = "degree", range = c(0.1, 0.9))
)
)Complete preprocessing pipeline
Baseline correction often works best as part of a complete preprocessing pipeline:
rec_complete <- recipe(water + fat + protein ~ ., data = meats) |>
step_measure_input_wide(starts_with("x_")) |>
# Baseline correction
step_measure_baseline_als(lambda = 1e6, p = 0.01) |>
# Scatter correction
step_measure_snv() |>
# Smoothing with mild derivative
step_measure_savitzky_golay(window_side = 5, differentiation_order = 1) |>
# Output for modeling
step_measure_output_wide(prefix = "nir_")
final_data <- bake(prep(rec_complete), new_data = NULL)
#> Warning: Values from `value` are not uniquely identified; output will contain list-cols.
#> • Use `values_fn = list` to suppress this warning.
#> • Use `values_fn = {summary_fun}` to summarise duplicates.
#> • Use the following dplyr code to identify duplicates.
#> {data} |>
#> dplyr::summarise(n = dplyr::n(), .by = c(water, fat, protein, location)) |>
#> dplyr::filter(n > 1L)
final_data[1:5, 1:8]
#> # A tibble: 5 × 8
#> water fat protein nir_01 nir_02 nir_03 nir_04 nir_05
#> <dbl> <dbl> <dbl> <list> <list> <list> <list> <list>
#> 1 60.5 22.5 16.7 <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]>
#> 2 46 40.1 13.5 <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]>
#> 3 71 8.4 20.5 <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]> <dbl [1]>
#> 4 72.8 5.9 20.7 <dbl [2]> <dbl [2]> <dbl [2]> <dbl [2]> <dbl [2]>
#> 5 58.3 25.5 15.5 <dbl [2]> <dbl [2]> <dbl [2]> <dbl [2]> <dbl [2]>Summary
-
ALS (
step_measure_baseline_als()) is a good default for most spectroscopy applications -
Polynomial
(
step_measure_baseline_poly()) works well for simple baseline shapes -
Derivatives via
step_measure_savitzky_golay()are effective for NIR/IR when peak shapes aren’t critical -
Custom functions
(
step_measure_baseline_custom()) provide maximum flexibility -
pybaselines
(
step_measure_baseline_py()) offers 50+ advanced algorithms when R options aren’t sufficient - Combine baseline correction with normalization (SNV/MSC) for best results